
| Макроцефалія | |
|---|---|
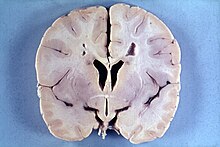
 МРТ пацієнта з доброякісною сімейною макроцефалією (чоловік з окружністю голови> 60 см)
| |
| Спеціальність | медична генетика |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LB70.3 |
| МКХ-10 | Q75.3 |
| OMIM | 248000 |
| DiseasesDB | 22519 |
| MeSH | C537717 |
| SNOMED CT | 9740002 |
| | |
Макроцефалія — це стан, при якому голова людини має аномально великий розмір; це стосується скальпу, черепу та його вмісту. Макроцефалія може бути патологічною, доброякісною, навіть генетично обумовленою. Людина з діагнозом макроцефалія має регулярно проходити перевірку, щоб визначити чи супроводжується вона якимись порушеннями.
Причини
Хоча в багатьох випадках макроцефалія не супроводжується неврологічними порушеннями, вона може бути патологічною. Патологічна макроцефалія обумовлюватися мегаленцефалією (збільшений мозок або за рахунок гіпертрофії та гіперплазії нервових елементів або за рахунок гіпертрофії й гіперплазії допоміжних клітин нервової тканини, що трапляється частіше), гідроцефалією (аномально підвищеною кількістю спинномозкової рідини), черепним гіперостозом (розростанням кісток) та іншими станами. Патологічну макроцефалію називають синдромічною, коли вона пов'язана з будь-яким іншим вартим уваги станом, і "несиндромною" в іншому випадку. Патологічна макроцефалія може зумовлюватися вродженими анатомічними порушеннями, генетичними станами або викликатися зовнішніми чинниками.
Макроцефалія, в тому числі сімейна, пов'язана з багатьма станами, зокрема з аутизмом; PTEN мутаціями, такими як хвороба Коудена, нейрофіброматозом типу 1 і туберозним склерозом; синдромами розростання, такими як синдром Сотоса (церебральний гігантизм), синдром Вівера, синдром Сімпсона-Голабі-Бемеля (синдром бульдога) та макроцефалічно-капілярною мальформацією ( M-CMTC); нейрокардіофаціально-шкірними синдромами, такими як синдром Нунана, синдром Костелло, синдром Горліна, та кардіофаціокутанозний синдром; Синдромом ламкої Х хромосоми; лейкодистрофіїями (дегенерації білої речовини головного мозку), такими як хвороба Александера, хвороба Канавана та мегаленцефальна лейкоенцефалопатія з кістами підкіркової тканини; і глутаровою ацидурією типу 1 і D-2-гідроксиглутаровою ацидурією.
Було виявлено, що хромосомні дуплікації пов'язані з аутизмом та макроцефалією; з іншого боку, виявлено, що хромосомні делеції пов'язані з шизофренією та мікроцефалією.
До зовнішніх чинників, пов’язаних з макроцефалією, належать інфекції, внутрішньошлуночкові кровотечі у новонароджених, субдуральні гематоми (кровотечі під зовнішньою оболонкою мозку), субдуральний випіт (збір рідини під зовнішньою оболонкою мозку) та арахноїдальні кісти (кісти на поверхні мозку),пухлини, черепно-мозкові травми.
При дослідженні висота черепа або візуалізація мозку можуть бути використані для більш точного визначення внутрішньочерепного об’єму.
Нижче наведені деякі причини макроцефалії.
Гідроцефалія







Гідроцефалія — захворювання, що характеризується надлишковим накопиченням цереброспінальної рідини у шлуночковій системі головного мозку в результаті ускладнення її переміщення від місця секреції (шлуночки головного мозку) до місця абсорбції до кровоносної системи (субарахноїдальних цистерн (субарахноїдального простору)) — окклюзійна гідроцефалія, або в результаті порушення абсорбції — арезорбти́вна гідроцефалія.

Причинами гідроцефалії, в свою чергу, можуть бути:
- мальформація Арнольда — Кіарі;
- акведуктальний стеноз;
- Х-зчеплена гідроцефалія зі стенозом водопроводу;
- синдром Денді — Уокера;
- аневризма вени Галена або мальформація;
- голопрозенцефалія;
- розширений субарахноїдальний простір(часто внаслідок крововиливу);
- внутрішньошлуночковий крововилив;
- саркоїдоз;
- менінгіт чи менінгоенцифаліт.
Супратенторіальна арахноїдальна кіста
Супратенторіальна арахноїдальна кіста — лікворна кіста, стінки якої сформовані клітинами павутинної оболонки. Є вродженою аномалією розвитку головного мозку, що може призводити до деформації черепу.
Менінгеальний фіброз/непрохідність
- постзапальний;
- постгеморагічний;
- пухлинна інфільтрація.

Судинні причини макроцефалії
- артеріовенозна мальформація;
- внутрішньочерепні крововиливи;
- тромбоз дурального синуса.
Папілома судинного сплетення
Нейрокутанні синдроми
- Інконтиненція пігментна
Деструктивні ураження
- гідранцефалія;
- поренцефалія.
Сімейні, аутосомно-домінантні, аутосомно-рецесивні, зчеплені з Х.

Причини макроцефалії пов'язані з субдуральною рідиною
Набряк мозку (токсично-метаболічний)
Зумовлений:
- інтоксикацією;
- свинцем;
- вітаміном А;
- тетрациклінами;
- ендокринними порушеннями(гіпопаратиреоз, гіпоадренокортицизм);
- галактоземією;
- ідіопатичний(внутрінньочерепна гіпертензія).

Потовщення черепу чи скальпу (гіперостоз)
- сімейна варіація;
- анемія;
- остеопороз, важкий аутосомно—рецесивний остеопороз(CLCN7, TCIRG1);
- пікнодизостоз(CTSK);
- краніометафізарна дисплазія(ANKH);
- краніодіафізарна дисплазія;
- пайлова дисплазія;
- склеростероз (SOST);
- ювенільна хвороба Педжета;
- ідіопатична гіперфосфатазія;
- сімейна остеоектазія;
- рахіт;
- клейдокраніальний дизостоз;
- синдром Ворта;
- синдром Протея.
Мегаленцефалія та гемімегаленцефалія
Мегаленцефалія — це порушення розвитку при якому мозок має аномально великий розмір. Середня вага мозку на 2,5 стандартні відхилення перевищує середнє значення загальної сукупності. Гемімегаленцефалія — надзвичайно рідкісна форма макроцефалії, що характеризується нерівномірним розвитком півкуль мозку (половина мозку більша за іншу).
Діагностика
Діагноз може бути встановлений внутрішньоутробно або протягом 18—24 місяців після народження. Діагностика у немовлят включає вимірювання окружності голови дитини та порівняння її з віковими нормами, перевищення цієї окружності на 2 стандартних відхилення від значення окружності голови для 97,5 процентиля дітей з подібними демографічними показниками може свідчити про макроцефалію. При значному перевищенні 97,5 процентиля, пацієнта перевіряють на наявність внутрішньочерепного тиску і визначають необхідність негайного хірургічного втручання. Якщо негайне оперативне втручання не потрібно, проводять подальші тестування, щоб визначити, чи є у пацієнта макроцефалія і чи доброякісна вона.
Доброякісна або сімейна макроцефалія
Доброякісна макроцефалія може виникати без причини або успадковуватися (при цьому вона вважається доброякісною сімейною макроцефалією і вважається мегаленцефальною формою макроцефалії). Діагноз сімейної макроцефалії визначається шляхом вимірювання окружності голови обох батьків та порівняння її з окружністю у дитини. Доброякісна та сімейна макроцефалія не пов'язані з неврологічними розладами, але нейророзвиток все одно оціненюється.
Хоча неврологічні розлади і не спостерігаються, можуть спостерігатися такі тимчасові симптоми, як: затримка розвитку, епілепсія та легка гіпотонія.
Лікування
Лікування варіюється залежно від наявності у дитини інших захворювань і того, де присутня спинномозкова рідина:
- якщо рідина виявлена між мозком та черепом, хірургічне втручання не потрібне;
- якщо між шлуночковими просторами в мозку виявляється надлишок рідини, знадобиться хірургічне втручання.
Асоційовані синдроми
Нижче наведено деякі синдроми, пов’язані з макроцефалією.

Пов'язані з чисельними аномаліями
- акрокалозальний синдром;
- синдром Аперта;
- Баннаян — Райлі — Рувалькаба;
- кардіофаціошкірний синдром;
- делеція хромосоми 22qter;
- клейдокраніальний дизостоз;
- синдром Костелло;
- енцефалокраніокутанний ліпоматоз;
- синдром FG;
- синдром Хайлермана — Стрейфа;
- гіпомеланоз Іто;
- синдром Лужана — Фрінса;
- синдром Маршалла — Сміта;
- нейрофіброматоз I;
- синдром Перлмана;
- синдром Рітшера — Шінцеля;
- синдром Робінова;
- синдром Сімпсона — Голабі — Бемеля;
- синдром Сотоса;
- синдром Стерджа — Вебера;
- синдром Вівера;
- синдром Відермана — Раутенштрауха.

Пов'язані з порушенням обміну речовин
- глутарова ацидурія II типу;
- гангліозидоз GM1;
- синдром Хантера;
- синдром Герлера;
- MPS VII;
- синдром Санфіліппо;
- синдром Зеллвегера.
Пов’язані зі скелетною дисплазією
- ахондроплазія;
- камптомелічна дисплазія;
- краниодіафізарна дисплазія;
- араниометалфізарна дисплазія;
- гіпохондрогенез;
- гіпохондроплазія;
- синдром Найста;
- остеопетроз, аутосомно—рецесивна форма;
- дисплазія Шнекенбекена;
- склеростеоз;
- синдром короткого ребра, тип Бемера — Лангера;
- вроджена спондилеопіфізарна дисплазія;
- танатофорна дисплазія.

Інші
- хвороба Вільяма Александера;
- хвороба Канавана;
- дефіцит кобаламіну(поєднана метилмалонова ацидурія та гомоцистинурія);
- синдром Денді — Вокера;
- глутарова ацидурія I;
- L-2 гідроксглутарова ацидурія;
- мегаленцефальна лейкоенцефалопатія;
- перивентрикулярна гетеротопія;
- хвороба Сандхофа;
- хвороба Тея — Сакса.