
| Синдром Ангельмана | |
|---|---|
 | |
| Спеціальність | медична генетика і неврологія |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | LD90.0 |
| МКХ-10 | Q93.5 |
| МКХ-9 | 759.89 |
| OMIM | 105830 |
| DiseasesDB | 712 |
| eMedicine | ped/1880 |
| MeSH | D017204 |
| | |
Синдром Ангельмана — обумовлена генетичною аномалією патологія, що характеризується такими ознаками як затримка психічного розвитку, порушення сну, напади, хаотичні рухи (особливо рук), частий сміх або посмішки. Також цю хворобу називають «синдромом Петрушки» або «синдромом щасливої ляльки».
При синдромі Ангельмана відсутні деякі гени довгого плеча 15-ї хромосоми (в більшості випадків спостерігається часткова делеція або інша мутація 15-ї хромосоми), причому страждає материнська хромосома, тоді як у разі пошкодження батьківської хромосоми виникає синдром Прадера-Віллі.
Каріотип 46 XX або XY, 15q-. Зазвичай синдром спричиняється спонтанним хромосомним дефектом, коли відсутня велика суміжна область з 3-4 мільйонів пар основ ДНК на ділянці q11-q13 15-ї хромосоми.
Згідно з результатами багатьох незалежних досліджень, причиною виникнення синдрому Ангельмана може бути мутація в гені UBE3A. Продукт цього гена — ферментний компонент складної системи деградації білків.
Синдром названий за ім'ям британського педіатра Гаррі Ангельмана, що вперше описав його в 1965 р в статті «Діти-„маріонетки“: Повідомлення про три випадки». Поштовхом до узагальнення — на основі типових клінічних проявів — тих випадків захворювання, які лікар спостерігав у своїй практиці, послужила побачена ним під час відпустки в Італії, в музеї Castelvecchio в Вероні картина Джованні Франческо Карото, на якій був зображений усміхнений хлопчик з малюнком шарнірної ляльки в руці.
Частота народження — від 1: 10 000 до 1: 20 000 живонароджених немовлят.
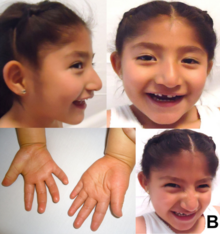
Особливості
Для синдрому Ангельмана характерні:
- У 75 % випадків — проблеми з харчуванням, особливо з грудним вигодовуванням, такі немовлята погано набирають вагу;
- Затримка в розвитку навичок загальної моторики (вміння сидіти, ходити);
- Затримка мовленнєвого розвитку, нерозвинене мовлення (у всіх дітей);
- Діти більше розуміють, ніж можуть сказати або висловити;
- Дефіцит уваги й гіперактивність;
- Складнощі при навчанні;
- Епілепсія (в 80 % випадків), порушення виявляються також при електроенцефалографії; вважається, що у дітей з синдромом Ангельмана спостерігається вторинна (симптоматична) епілепсія;
- Незвичайні рухи (дрібний тремор, хаотичні рухи кінцівок);
- Частий сміх без приводу;
- Ходьба на негнучких ногах — через цю особливість дітей із цим синдромом іноді порівнювали з маріонетками;
- Розмір голови менший від середнього, нерідко зі сплощенням потилиці;
- Іноді характерні риси обличчя — широкий рот, рідко розташовані зуби, висунуті вперед підборіддя та язик;
- Розлади сну;
- Косоокість в 40 % випадків;
- Сколіоз (викривлення хребта) в 10 % випадків;
- Підвищена чутливість до високої температури;
- Відчувають себе більш комфортно у воді.
Діагностика
Синдром діагностується шляхом генетичного аналізу (15а хромосома), рекомендованого для новонароджених зі зниженим м'язовим тонусом (гіпотонусом), відставанням у розвитку загальної моторики і в розвитку мови. Батьки і лікарі повинні звернути увагу на випадки дрібного тремору, хаотичні, рвучкі рухи кінцівок, ходу з негнучкими ногами; в ряді випадків специфічний вираз обличчя, занадто частий сміх.
Можливі методи аналізу: процес флуоресцентної гібридизації in situ, метилювання ДНК в області 15q11-q13, аналіз мутації імпринтингового центру, аналіз прямої мутації гена UBE3A.
Лікування
Синдром Ангельмана обумовлений вродженою генетичною аномалією; специфічні способи його лікування наразі не розроблені, проте деякі лікувальні заходи підвищують якість життя людей із синдромом. Немовлята з гіпотонусом повинні отримувати масаж і інші види спеціальної терапії (фізіотерапії).
Рекомендуються використання спеціальних методик розвитку дитини, заняття з логопедом і дефектологом. Порушення сну коригуються призначенням легких снодійних. Доктор Вагстафф (США) вважає, що призначення 0,3 мг мелатоніну за 0,5-1,0 год перед сном покращує сон пацієнтів з синдромом Ангельмана. Порушення стільця регулюються призначенням легких проносних.
При судомних нападах підхід до лікування такий самий, як при епілепсії. У дітей з синдромом Ангельмана часто спостерігаються напади більше ніж одного типу. Показана електроенцефалографія.
Вимагає корекції небажаної поведінки. Д-р Чарльз Вільямс (Гейнсвілл, Флорида), що працює в основному з аутичними дітьми, зазначає спільні для аутичних дітей і дітей з синдромом Ангельмана особливості поведінки: помітні аутостимуляція, імпульсивність, нав'язливі, повторювані рухи, інтерес до недоречних предметів, а також складнощі при спілкуванні з іншими людьми. Лікарі США повідомляють, що у аутичних дітей внутрішньовенні ін'єкції гормону «секретин» (знайденого в підшлунковій залозі) успішно зменшують прояви небажаної поведінки і забезпечують хороший рівень товариськості і комунікативних навичок; можливо, медицина прийде до використання секретину для корекції поведінки дітей з синдромом Ангельмана.
Ризики
Оцінка ризику повторного народження дитини з синдромом Ангельмана у тих самих батьків дуже складна, необхідна консультація професійного генетика. Вважається, що звичайна делеція є спонтанною, ризик повторення менший, ніж 1 %. У разі молекулярної мікроделеції в ділянці 15q11-q13, якщо вона спостерігається у матері, ризик теоретично зростає до 50 %. Мутації всередині гена UBE3A можуть бути випадковими і неуспадкованими, в цій ситуації ризик повторення <1 %; однак ці мутації можна успадкувати від матері без мутації, і тоді теоретичний ризик — 50 %. Партеногенетична дисомія 15-й пари — випадкова ситуація; ризик повторення <1 %. У рідкісних випадках синдрому Ангельмана з незвичайними перетвореннями 15-ї хромосоми, включаючи ділянку 15q11-q13, оцінка ризику повтору залежить від хромосомних порушень у батьків.
Перспективи розвитку
Діти з синдромом Ангельмана розуміють набагато більше, ніж можуть сказати. У деяких випадках у них взагалі немає мови; є описані випадки — діти зі словниковим запасом в 5-10 слів. При цьому діти з синдромом Ангельмана люблять спілкуватися з іншими людьми, грати, як правило, вони доброзичливі.
Рекомендується навчати таких дітей мови жестів. Заняття з раннього віку за спеціальними програмами, спрямовані на розвиток навичок дрібної і загальної моторики, в ряді випадків дають хороші результати.
Перспективи розвитку залежать від ступеня ураженості хромосоми. Деякі люди з синдромом Ангельмана здатні освоїти навички самообслуговування і мову на примітивному рівні (зазвичай причиною синдрому в цьому випадку стає мутація), деякі ніколи не зможуть ходити і говорити (таке зазвичай відбувається в разі делеції частини хромосоми).
З віком, як правило, симптоми гіперактивності та порушення сну пом'якшуються. У дівчаток з синдромом Ангельмана в період статевого дозрівання можуть почастішати напади.
Більшість людей з синдромом Ангельмана здатні контролювати екскреторні функції (сечовипускання і дефекацію) вдень, деякі і вночі. Частина людей з синдромом Ангельмана здатні їсти за допомогою ножа і виделки, одягатися самостійно (при відсутності на одязі ґудзиків, «блискавок» і т. ін.). У дорослому віці може з'явитися ожиріння і погіршитися ситуація зі сколіозом.
Статеве дозрівання індивідів з синдромом Ангельмана, зокрема поява менархе у дівчаток, відбувається в звичайні терміни. Описано один випадок вагітності жінки з синдромом Ангельмана: вона народила дівчинку з таким самим діагнозом.
Посилання
- Angelman Syndrome Foundation [Архівовано 4 травня 2012 у Wayback Machine.]
- Angelman syndrome (англ.). U.S. National Library of Medicine. [Архівовано 27 серпня 2016 у Wayback Machine.]
- Angelman syndrome; AS.. Happy puppet syndrome, formerly [Архівовано 29 травня 2014 у Wayback Machine.] (англ.). Online Mendelian Inheritance in Man.
|