
| Хвороба Гоше | |
|---|---|
 Кисла β-глюкозидаза
| |
| Спеціальність | ендокринологія і неврологія |
| Препарати | Циклосерин, Велаглюцераза альфа, Іміглюцераза, eliglustatd і miglustatd |
| Частота | 1:40 000 |
| Класифікація та зовнішні ресурси | |
| МКХ-11 | 5C56.0Y |
| МКХ-10 | E75.2 |
| OMIM | 230800 |
| DiseasesDB | 5124 |
| MedlinePlus | 000564 |
| MeSH | D005776 |
Хвороба Гоше або Сфінголіпідоз (ХГ) — рідкісне спадкове захворювання, яке викликане генетичними мутаціями та успадковується за аутосомно-рецесивним типом. Ці мутації обумовлюють дефіцит активності лізосомального ферменту глюкоцереброзидази (також відомий як люкозилцерамідаза), внаслідок чого порушується лізосомальне зберігання. Це призводить до накопичення субстрату (глюкоцереброзиду) в лізосомах макрофагів та інших клітин, наприклад, остеобластів. Заповнені ліпідами «клітини Гоше» акумулюються в різних тканинах і органах, особливо в селезінці, печінці, кістковому мозку, легенях та мозку.
Прояви можуть включати збільшення селезінки та печінки, порушення функції печінки, скелетні розлади або ураження кісток, що бувають досить болючими, серйозні неврологічні ускладнення, набряк лімфатичних вузлів та іноді, прилеглих до них суглобів, здуття живота, буруватий відтінок шкіри, анемію, низький рівень тромбоцитів у крові та жирові відкладення жовтого кольору на склері ока. Пацієнти з важкими формами можуть мати підвищенну вразливість до інфекційних захворювань. Деякі форми хвороби Гоше можна лікувати за допомогою ферментозамісної терапії.
Серед населення Західної та Східної Європаи частота Хвороби Гоше становить від 1:40 000 до 1:60 000. Нейропатичний тип ХГ зустрічається значно рідше і становить від 1:50 000 до 1:100 000 населення. Особиво поширеним захворювання є у популяції євреїв-ашкеназі, де частота досягає 1:850. Станом на 2022 рік в Україні живуть 67 пацієнтів з діагностованою хворобою Гоше.
Хвороба Гоше є найпоширенішою з лізосомальних хвороб накопичення. Є формою сфінголіпідозу (підгрупа лізосомальних хвороб накопичення), оскільки пердбачає дисфункціональний метаболізм сфінголіпідів.
Симптоми та ознаки
- Затримка фізичного та статевого розвитку;
- Слабкість, підвищена втомлюваність (астенія);
- Часті респіраторні інфекції;
- Ознаки геморагічного синдрому (підшкірні гематоми, підвищена кровоточивість слизових оболонок, тривалі кровотечі при малих оперативних втручаннях);
- Біль у кістках та суглобах, часті переломи кісток;
- Остеопороз: у 75 % пацієнтів розвиваються видимі аномалії кісток через накопичення в них глюкозилцераміду. Зазвичай описують деформацію дистального відділу стегнової кістки у формі колби Ерленмейера.
- Неврологічна симптоматика (окорухова апраксія або збіжна косоокість, атаксія, зниження інтелекту, порушення чутливості);
- Безболісна гепатоспленомегалія (збільшення печінки та селезінки). Розмір селезінки може досягати 1500-3000 г, тоді як нормальний розмір — 50-200 г. Спленомегалія може зменшити здатність ураженої особи приймати їжу через тиск на шлунок. Хоча збільшення селезінки є безболісним, воно збільшує ризик її розриву, що призводить до важких ускладнень.
- Лімфаденопатія, гіперпігментація шкіри в області ліктьових та колінних суглобів.
- Фактором ризику є наявність у сімейному анамнезі спленектомії або інших наведених вище симптомів у рідних братів чи сестер;
- Встановлено, що у пацієнтів із хворобою Гоше та їхніх гетерозиготних родичів-носіїв частіше зустрічається хвороба Паркінсона.
Серед перелічених ознак у пацієнтів із ХГ найчастіше виявляють гепатомегалію (87 %), спленомегалію (95 %), анемію (40 %), тромбоцитопенію (50 %), колби Ерленмеєра (49 %), інфільтрацію кісткового мозку (39 %), біль у кістках (27 %), остеопенію (20 %), затримку росту (<5 перцентиля — 28 % і в межах 5-25 перцентиля — 28 %).
Генетика
Усі типи хвороби Гоше передаються за автосомно-рецесивним типом. Для народження хворої дитини, обоє батьків повинні бути носіями. Якщо обидва батьки є носіями, ймовірність народження хворої дитини становить 1 до 3. Попередження народження дітей з ХГ полягає у проведенні медикогенетичного консультування з подальшою преконцепційною підготовкою та/або пренатальною діагностикою у сім'ях групи високого ризику захворювання.
Останні десятиріччя проводиться ретельний аналіз мутацій пов'язаних з хворобою Гоше. Хоча і було знайдено близько 300 мутацій гена GBA1, на переважну більшість пацієнтів припадає лише декілька з них. Вважається, що 4 мутації (N370S, IVS2 (+1), 84GG, L444P) зустрічаються у 96,5 % хворих ашкеназьких євреїв в західній півкулі та у 50–60 % пацієнтів неєврейської популяції. Наявність як мінімум однієї алелі з мутацією N370S виключає розвиток нейропатичного захворювання, а наявність L444P тісно пов'язана (хоча не винятково) із залученням нервової системи.
За наявністю деяких мутацій виділяють такі типи ХГ:
- Тип I (гомозиготна N370S) — найпоширеніший, також званий «ненейропатичним» типом. Дуже часто зустрічається у євреїв-ашкеназі, у 100 разів частіше, ніж серед загального населення. Середній вік на момент встановлення діагнозу становить 28 років, а очікувана тривалість життя дещо зменшена.
- Тип II (одна або дві алелі L444P) — характеризується гострими неврологічними проблемами у маленьких дітей. Фермент майже не виділяється в лізосоми. Прогноз поганий: більшість хворих помирає у віці до трьох років. Не має жодної специфіки для жодної етнічної групи.
- Тип III (також одна або дві копії L444P, проте, можливо, затримані захисними поліморфізмами) зустрічається у шведських пацієнтів із регіону Норрботтен, серед арабів та у країнах Азії. У цієї групи хвороба розвивається дещо пізніше, але більшість помирає до свого 30-річчя.
Патофізіологія
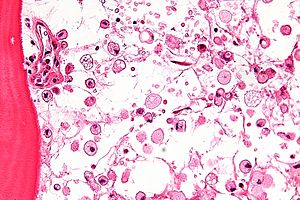
Хвороба спричинена дефектом конститутивного гена для лізосомальної глюкоцереброзидази, також відомої як кисла β-глюкозидаза, (КФ 3.2.1.45, PDB 1OGS) на першій хромосомі (1q22). Фермент являє собою білок вагою 55,6 кілодальтон і довжиною 497 амінокислот, який каталізує гідроліз глюкоцереброзиду, компонента клітинної мембрани червоних і білих кров'яних тілець. При хворобі Гоше фермент не може функціонувати належним чином, внаслідок мутацій, і глюкоцереброзид накопичується. Макрофаги, що поглинають ці клітини, не можуть метаболізувати накопичені ліпіди, і перетворюються на «клітини Гоше», які під світловим мікроскопом виглядають як зім'ятий папір.
Різні типи мутацій в гені GBA1 (β-глюкозидази) визначають різну залишкову активність ферменту. При I типі спостерігається деяка залишкова активність ферменту, що запобігає розвитку нейропатологій при цьому типі. Хоча й існує деяка кореляція між генотипом і фенотипом захворювання, проте, ані кількість накопичених ліпідів, ані рівень залишкової активності ферменту не корелюють напряму із симптомами захворювання та їх важкістю. Цей факт зумовлює необхідність подальшого дослідження деяких симптомів, як от:
- блокування ендо/лізосомальної системи
- Стрес ендоплазматичного ретикулуму
- змінений ліпідний склад мембран по всій клітині, включаючи плазматичну мембрану, і наступні зміни в динамічних і сигнальних властивостях клітинної мембрани
- запалення, викликане секрецією цитокінів в результаті накопичення сфінголіпідів, та нейродегенерація внаслідок накопичення глюкозилсфінгозину, що є нейротоксином
Гетерозиготні варіани для певних мутацій кислої β-глюкозидази півищують ризик розвитку хвороби Паркінсона приблизно у п'ять разів, що робить ХГ найпоширенішим відомим генетичним фактором ризику хвороби Паркінсона.
Також хворобу Гоше пов'язують з підвищенним ризиком розвитку раку, особливо мієломи. Вважається, що це відбувається через накопичення у клітинах глюкозилцераміду та складних глікосфінголіпідів.
Роль запальних процесів при хворобі Гоше вивчена недостатньо. Однак відомо, що сфінголіпіди беруть участь у запаленні та апоптозі. Також, у людей з хворобою Гоше спостерігається підвищений рівень маркерів активації макрофагів. До таких факторів відносять ангіотензинперетворюючий фермент, катепсин S, хітотріозидазу та CCL18 у плазмі крові; та фактор некрозу пухлини альфа в клітинах Гоше в селезінці.
Класифікація
За рівнем залучення нервової системи розрізняють три загальні клінічні підтипи хвороби Гоше (ХГ). Втім цей поділ зазнав певної критики через те, що він не охоплює повний спектр можливих симптомів (фенотипів). Також виникають складні гетерозиготні варіації, що значно ускладнює прогнозування перебігу захворювання.
ХГ I типу (ненейропатична, або вісцеральна форма, OMIM 230800) є найпоширенішою і найменш важкою формою захворювання. Симптоми можуть виникати у дитячому або зрілому віці і головним чином вражати печінку, селезінку та кістки. Часто спостерігаються збільшення печінки та значне збільшення селезінки (разом гепатоспленомегалія); селезінка може розірватися і викликати додаткові ускладнення. Може спостерігатися значна слабкість скелета та важкі захворювання кісток. Збільшення селезінки та заміщення кісткового мозку викликають анемію, тромбоцитопенію та лейкопенію. Мозок і нервова система патологічно не уражені, але можуть виникнути ураження легенів і, рідко, нирок. У пацієнтів цієї групи зазвичай легко утворюються синці (через низький рівень тромбоцитів), вони схильні швидко втомлюватись через низьку кількість еритроцитів. Залежно від часу початку захворювання та його тяжкості, пацієнти типу I можуть доживати до зрілого віку. Симптоми та їх важкість може різко варіюватись серед пацієнтів.
ХГ II типу (гостра нейропатична форма, OMIM 230900) зазвичай проявляється протягом 6 місяців після народження. Частота захворюваності — близько 1 на 100 000 живонароджених. Симптоми включають збільшення печінки та селезінки, обширне та прогресуюче ураження головного мозку, порушення рухів очей, спастичність, судоми, ригідність кінцівок та погану здатність смоктати та ковтати. Хворі діти зазвичай помирають у віці до двох років.
ХГ III типу (хронічна невропатична, OMIM 231000) може проявлятись як у дитинстві, так і, іноді, дорослому віці та зустрічається приблизно у одного зі 100 000 живонароджених. Вона характеризується неврологічними симптомами більш м'якими порівняно з типом II, які, проте, повільно прогресують. Основні симптоми включають збільшення селезінки та/або печінки, судоми, погану координацію, порушення скелета, порушення рухів очей, захворювання крові, включаючи анемію, і проблеми з диханням. Пацієнти часто можуть доживати до підліткового, або навіть зрілого віку. Існує дві рідкісні форми хвороби: підтипи ІІІ типу — IIIb та ІІІс. IIIb поширена у Північній Швеції та характеризується переважанням вісцеральних проявів над невропатичними. При захворюванні з субтипом ІІІс відбувається ураження серцевих клапанів — цей субтип переважає серед палестинських арабів.
Діагностика
Хворобу Гоше припускають на підставі загальної клінічної картини. Початкове лабораторне дослідження може включати дослідження ферментів. У результаті активність ферменту менша за 15 % середньої активності у нормі вважається діагностичностичною.
При підозрі на хворобу Гоше для встановлення остаточного діагнозу слід провести такі дослідження:
- вимірювання активності глюкоцереброзидази в сухих плямах крові як скринінг-метод, який дозволяє зменшити строки діагностики ХГ;
- вимірювання активності глюкоцереброзидази у лейкоцитах крові, що є високоточним методом, який дозволяє встановити діагноз ХГ;
- вимірювання активності хітотриозидази є допоміжним тестом при діагностиці ХГ, оскільки значна частка населення є носіями алелей, що призводять до низької активності цього ферменту. Крім того, активність хітотриозидази є важливим маркером успішності лікування ХГ;
- молекулярно-генетична діагностика з метою виявлення мутацій у гені глюкоцереброзидази.
Лікування
Для пацієнтів із типом I та більшою частиною типів III ферментозамісна терапія внутрішньовенною рекомбінантною глюкоцереброзидазою може зменшити розмір печінки та селезінки, заповільнити розвиток аномалій скелета та зняти інші симптоми. Таке лікування коштує приблизно 200 000 доларів США на рік для однієї людини і повинно тривати довічно. У зв'язку з рідкістю даного захворювання, досілідження необхідних терапевтичних дозуваннь є дуже складним, тому щодо цього питання досі точаться дискусії Через низьку поширеність звороби Гоше, у багатьох країнах цей препарат визнаний орфанним препаратом, що означає, що уряди фінансово компенсують ринкову недоцільність дослідження та виробництва таких препаратів, пов'язану з маленьким попитом на них.
Першим препаратом для лікування хвороби Гоше була альглюцераза (Ceredase), яка була версією глюкоцереброзидази, яку збирали з плацентарної тканини людини, а потім модифікували ферментами. Він був схвалений FDA у 1991 році та був пізніше вилучений з ринку через схвалення подібних препаратів, виготовлених за технологією рекомбінантної ДНК замість виділення з тканини. Пепарати, отримані рекомбінантним шляхом, є кращими, оскільки у разі їх використання відсутня небезпека інфекційних хвороб, джерелом яких можуть бути тканини, які використовуються для виділення ферменту, а також забезпечується стабільніша структура ферменту від партії до партії. Окрім того вони дешевші у виробництві.
Сьогодні в Україні зареєстровані такі препарати для ферментозамісної терапії:
- Таліглюцераза (Елелісо, Pfizer) — рекомбінантний аналог лізосомальної глюкоцереброзидази людини, виробляється за технологією рекомбінантної ДНК із використанням культури рослинних клітин (моркви);
- Велаглюцераза альфа — виготовляється за допомогою людської лінії фібробластів; амінокислотна послідовність у препараті ідентична ендогенній глюкоцереброзидазі;
- Іміглюцераза — виготовляється клітинною лінією, отримана з яєчників китайських хом’яків.
Крім основного лікування, хворому на ХГ призначається симптоматичне лікування (при остеопорозі — дієта, збагачена кальцієм, вітаміном D, при кістковому кризі — аналгетики, при бактеріальній інфекції — антибактеріальну терапію, ортопедичне лікування, при неврологічній симптоматиці — ноотропи, протиепілептичні препарати, міорелаксанти тощо). Також при веденні пацієнта з ХГ важливе значення має профілактика сепсису при функціональному гіпоспленізмі, основним напрямком якої є вакцинація проти пневмококової, менінгококової та Hib-інфекції.

Історія
Хвороба була вперше розпізнана французьким лікарем Філіпом Гоше, який описав її в 1882 році та дав назву. У 1902 році Натан Бріл відкрив спосіб її успадкування. Пошкодження нейронів, пов'язане з цією хворобою, було виявлено в 1920-х роках, а біохімічні основи хвороби були з'ясовані в 1960-х роках Роско Брейді. У квітні 1991 року FDA схвалила перше ефективне лікування захворювання — препарат альглюцеразу (Ceredase). Покращений препарат, іміглюцераза (Cerezyme), був схвалений FDA в травні 1994 року і замінив використання Ceredase.